SINDROMUL TURNER
Incidenta
la nastere, este, conform majoritati statisticilor, de 0,4.
Incidenta produsilor de conceptie care prezinta
aceasta anomalie este mult mai mare dar, aproximativ 98% din fetii cu
cariotip 45, XO sunt avortati spontan.
Amenoreea primara reprezinta regula, desi
au fost citate cateva paciente care au prezentat menstruatie
intermitenta sau, in mod exceptional regulata. Asemenea cazuri
trebuie sa evoce prezenta unui mozaic, mai ales atunci cand sunt
insotite de fertilitate. Ovarele sunt lipsite de foliculi fiind
reprezentate de doua bandalete fibroase, in interesul carora se poate
recunoaste adesea, aspectul stromei ovariene. In decursul dezvoltarii
embrionare ovarele contin un numar normal de foliculi care ulterior
dispar datorita unui proces de involutie progresiva.
Insuficienta ovariana periferica din sindromul Turner se traduce
prin concentratii serice foarte reduse alee hormonilor estrogeni si
progesteronului (secretati aproape exclusiv de corticosuprarenala)
si eliminari urinare foarte reduse ale metabolitilor acestora
(fenolsteroizi si pregnaudiol). Concentratia serica a hormonilor
gonadotropi, LH, FSH are valori mult crescute. Cresterea constanta a
acestora si raspunsul pozitiv la stimularea cu
'releasing-hormon' (LH-RH) este considerata cea mai
importanta determinare hormonala in sindromul Turner.
Trompele si mai ales uterul
au un aspect infantil, hipoplazic. Glandele mamare sunt putin dezvoltate,
areolele sunt mici si nepigmentate, iar distanta intermamelonara
este crescuta. Pilozitatea axilara si pubiana este
redusa sau absenta. Vulva are un caracter infantil, labiile mari sunt
putin dezvoltate si nepigmentate, iar labiile mici sunt adesea
inaparente. Clitorisul este de obicei normal. Uneori se intalneste o
hipertrofie clitoridiana moderata, expresie a unui anumit grad de
virilizare. Excretia de 17-cetosteroizi este scazuta, in timp ce
nivelul gonadotropinelor este crescut.
Greutatea la nastere se
incadreaza in limitele prematuritatii iar inaltimea
femeilor adulte nu depaseste 150 cm. Anomaliile scheletice
includ: retrognatie si micrognatie, aspectul bombat al
toracelui, cresterea diametrului biacromial care depaseste
diametrul bitrochanterian, anomalii vertebrale, cu malformatii ale
corpilor vertebrali sau fuziunea vertebrelor cervicale, condrodistrofia
platoului tibial si a condilului intern (semnul Kosowicz);
brachimetacarpalism (scutarea metacarpienelor, cu deosebire a celor de pe
partea cubitala (semnul Archibald), anomalii ale degetelor mainii,
constand in camptodactilie (flexiunea falangelor) si sindactilie;
osteoporoza difuza sau localizata la bazin, coloana
vertebrala, oasele tarsului sau corpului.
La
nou-nascuti se observa, in 15-20% din cazuri un exces de
tegument in regiunea cefei si limfedeme periferice. La adulti se
remarca prezenta unor pliuri cutanate situate intre mastoida
si acromion, care dau gatului un aspect scurt, foarte caracteristic,
accentuat de implantarea joasa a parului, in regiunea cefei.
Pielea
prezinta nevi pigmentari sau papiloame. Unghiile, hipoplazice, sunt scurte
si patrate. Cea mai frecventa boala cardiaca
congenitala este coarctatia aortei, mai rar se observa
comunicatiile interatriale. Anomaliile renale sunt destul de frecvente
si constau in rinichi in potcoava si ureter dublu. Exista
adesea tulburari auditive. In ordinea frecventei, anomaliile oculare
constau in: epicantus, strabism, ptoza palpebrala, cataracta,
hipertelorism si nistagmus.
Nivelul
hormonilor de crestere hipofizari poate atinge nivele tot atat de crescute
ca in acromegalie, ceea ce sugereaza o insensibilitate a organelor
tinta. Procesele autoimune si indeosebi producerea de anticorpi
antitiroglobulinici, par destul de frecvente.
Nivelul
dezvoltarii intelectuale este normal sau usor redus. Spre deosebire
de barbatii cu sindrom Klinefelter si de femeile cu tripliodie
X, femeile cu sindrom Turner nu sunt intalnite cu o frecventa
crescuta in grupa pacientelor cu debilitate mintala grava.
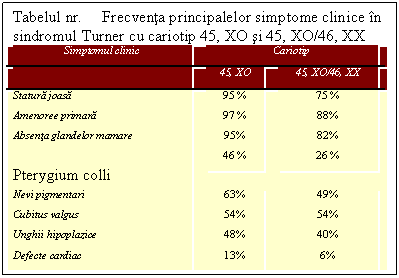 Pacientele
cu sindrom Turner pot prezenta doua sau mai multe populatii celulare
diferite, una dintre ele fiind linia XO. In 10% din cazurile de sindrom Turner,
alaturi de linia XO se gaseste o linie celulara
normala XX. Acest tip de mozaicism apare prin pierderea unui cromozom X
intr-un stadiu precoce de clivare a embrionului. Fenotipul acestor cazuri este
extrem de variat de la cel de Turner complet pana la absenta totala
a anomaliilor si fertilitate normala. Acest spectru larg al
manifestarilor clinice este determinat de proportia diferita in
care se pot gasi cele doua linii celulare. Spre deosebire de
pacientele cu sindromul Turner XO, care prezinta cu predilectie
coarctatia aortei, pacientele cu mozaic XO/XX prezinta stenoza
aortica, cu sau fara defecte septale atriale.
Pacientele
cu sindrom Turner pot prezenta doua sau mai multe populatii celulare
diferite, una dintre ele fiind linia XO. In 10% din cazurile de sindrom Turner,
alaturi de linia XO se gaseste o linie celulara
normala XX. Acest tip de mozaicism apare prin pierderea unui cromozom X
intr-un stadiu precoce de clivare a embrionului. Fenotipul acestor cazuri este
extrem de variat de la cel de Turner complet pana la absenta totala
a anomaliilor si fertilitate normala. Acest spectru larg al
manifestarilor clinice este determinat de proportia diferita in
care se pot gasi cele doua linii celulare. Spre deosebire de
pacientele cu sindromul Turner XO, care prezinta cu predilectie
coarctatia aortei, pacientele cu mozaic XO/XX prezinta stenoza
aortica, cu sau fara defecte septale atriale.
Au fost descrise si cateva cazuri de sindrom
Turner cu cariotip normal 46,XX, dar realitatea lor este discutabila. Este
vorba, probabil, de mozaicuri in care linia XO este nedetectata, deoarece
proportia in care se gaseste este foarte mica sau deoarece
prezenta sa este limitata la anumite teritorii.
Aproximativ 15-20% din
pacientele cu sindrom Turner prezinta un izocromozom X. Studiul grupelor
sangvine Xq sugereaza ca izocromozomul X se formeaza in decursul
meiozei masculine. Majoritatea pacientelor cu izocromozom X sunt mozaicuri
XO/X, i (Xq). Prezenta liniei XO se explica prin retardarea
anafazica si pierderea izocromozomului din celulele zigotului. Au
fost descrise cariotipuri mai complexe 46, X, i (Xq)/47, X, i (Xp), Xp- sau
46,X, inv / (Xq) dar frecventa lor este foarte redusa.
Incidenta
corpusculilor Barr este destul de mica, dar nu sunt neobisnuiti.
Simptomatologia clinica (statura joasa, infantilism sexual,
amenoree primara, anomalii scheletice) este asemanatoare cu cea
a sindromului Turner XO.
Diagnosticul se stabileste
de obicei in perioada pubertatii, cand absenta caracteristicilor
secundare si mai ales amenoreea reprezinta un motiv de neliniste
majora in familia pacientelor si impun solicitarea consultului
medical. De cele mai multe ori pare inexplicabil faptul ca s-a facut
abstractie de perturbarea grava a ritmului de crestere, de
deficitul in dezvoltarea intelectuala si de dismorfismul craniofacial
evident. Uneori in absenta principalelor caracteristici sexuale ca telarha
si purbaha incompleta, singurul semnal de alarma il
reprezinta amenoreea la varsta de 14-16 ani. Variabilitatea varstei la
care se solicita consultul medical pentru amenoree se coreleaza cu
mediul de provenienta socio-economica si cu gradul de
scolarizare a mamei.
Contextul clinic, cariograma
si determinarile hormonale specifice permit diagnosticul
diferential in majoritatea acestor cazuri.

